



یماریهای متابولیک نادر مانند بیماریهای ذخیرهای لیزوزومی و اختلالات میتوکندریایی در نگاه نخست ممکن است ناشناخته یا پیچیده به نظر برسند، اما آگاهی دقیق درباره آنها میتواند به تشخیص زودهنگام، مدیریت مناسب و بهبود کیفیت زندگی بیماران کمک کند. این بیماریها، اگرچه اغلب ارثی هستند، با پیشرفت علم قابل تشخیص دقیق و در بسیاری موارد قابل کنترل میباشند.
بیماریهای ذخیرهای لیزوزومی چیست؟
لیزوزومها اندامکهای کوچک درون سلول هستند که با استفاده از آنزیمها، مواد زائد و مولکولهای پیچیده را تجزیه کرده و به اجزای قابل استفاده یا دفع تبدیل میکنند.
در بیماریهای ذخیرهای لیزوزومی (Lysosomal Storage Diseases – LSDs):
– نقص یا کمبود یک آنزیم خاص در لیزوزوم رخ میدهد.
– مواد غیرقابل تجزیه (مانند چربیها، قندهای پیچیده یا گلیکوزآمینوگلیکانها) در سلولها تجمع میکنند.
– به مرور زمان این تجمع باعث آسیب به بافتها و اندامهای مهمی مثل کبد، طحال، قلب، استخوانها و سیستم عصبی میشود.
نمونههایی از بیماریهای لیزوزومی:
- بیماری گوشر (Gaucher)
- بیماری فابری (Fabry)
- بیماری نیمن–پیک (Niemann–Pick)
- موکوپلیساکاریدوزها (MPS)
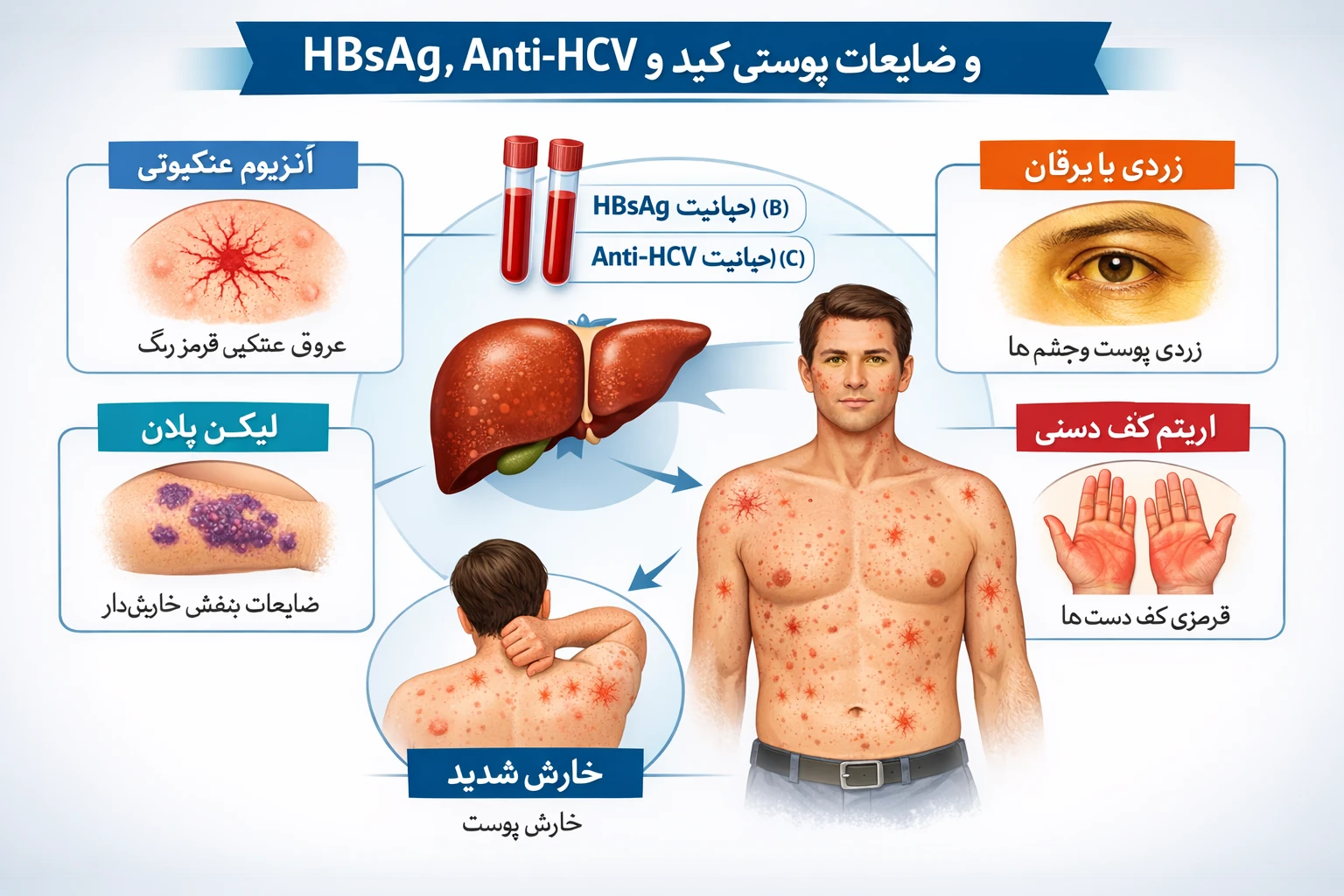
علائم شایع بیماریهای ذخیرهای لیزوزومی
– بزرگشدن کبد و طحال (هپاتواسپلنومگالی)
– مشکلات رشد و کوتاهی قد
– اختلال در یادگیری یا عملکرد ذهنی
– تغییرات بینایی یا شنوایی
– درد و ضعف عضلانی
– تغییرات اسکلتی
اختلالات میتوکندریایی چیست؟
میتوکندری، «نیروگاه سلول»، انرژی مورد نیاز بدن را به شکل ATP تولید میکند. در اختلالات میتوکندریایی:
– نقص ژنتیکی در DNA میتوکندری یا هسته سلول باعث اختلال در زنجیره تنفسی سلولی میشود.
– سلولها انرژی کافی تولید نمیکنند که بیشترین آسیب را به اندامهای پرانرژی مانند مغز، عضلات، قلب و کلیهها میزند.
نمونههایی از اختلالات میتوکندریایی:
– MELAS (انسفالوپاتی میتوکندریایی همراه با اسیدوز لاکتیک و اپیزودهای سکتهمانند)
– MERRF (صرع میوکلونیک با فیبرهای رگبهرگ قرمز)
– سندرم لی (Leigh syndrom
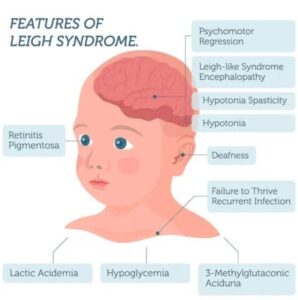
علائم شایع اختلالات میتوکندریایی
– خستگی مزمن و عدم تحمل فعالیت
– ضعف و گرفتگی عضلات
– تأخیر رشد حرکتی یا شناختی
– اختلالات بینایی و شنوایی
– مشکلات قلبی (کاردیومیوپاتی)
– تشنج و سکتههای کوچک در سنین جوانی
روشهای تشخیص
برای هر دو گروه بیماری، ترکیبی از روشهای زیر استفاده میشود:
– آزمایش خون و ادرار برای بررسی مواد ذخیرهشده یا اختلالات متابولیکی
– آنالیز آنزیمی (برای LSDs)
– بررسی DNA و تستهای ژنتیک
– نمونهبرداری بافتی (بیوپسی عضله یا کبد)
– تصویربرداری MRI یا CT برای بررسی آسیب عصبی یا اندامی
درمان و مدیریت بیماریها:
باوجود اینکه بسیاری از این بیماریها درمان قطعی ندارند، مدیریت آنها پیشرفت زیادی داشته است:
– درمان جایگزینی آنزیم (ERT): برای برخی بیماریهای لیزوزومی مانند گوشر و فابری
– درمان کاهش بستر (Substrate Reduction Therapy): کاهش تولید مواد تجمعپذیر
– مکملهای غذایی و کوفاکتورهای متابولیکی: در برخی اختلالات میتوکندریایی (مانند کوآنزیم Q10)
– فیزیوتراپی و کاردرمانی برای بهبود حرکت و کیفیت زندگی
– کنترل علائم مانند داروهای ضدتشنج یا درمان مشکلات قلبی
پیشآگهی و اهمیت تشخیص زودهنگام
تشخیص زودهنگام نهتنها میتواند شروع درمان را تسریع کند، بلکه امکان کاهش پیشرفت آسیب اندامی و بهبود عملکرد بیمار را فراهم میکند. مشاوره ژنتیک برای خانوادههایی که سابقه این بیماریها را دارند، بسیار توصیه میشود.
پرسشهای متداول
۱. آیا این بیماریها ارثی هستند؟
بله، اغلب این بیماریها به صورت ارثی (اتوزومال مغلوب یا وابسته به X) منتقل میشوند.
۲. آیا درمان قطعی وجود دارد؟
برای بسیاری از موارد درمان قطعی وجود ندارد، اما با درمانهای جدید میتوان علائم را کنترل و روند بیماری را کند کرد.
۳. آیا سبک زندگی بر پیشرفت بیماری اثر دارد؟
در برخی موارد، رعایت رژیم غذایی مناسب، کنترل استرس و پیروی از برنامه ورزشی سبک میتواند به بهبود کیفیت زندگی کمک کند.
جمعبندی
بیماریهای ذخیرهای لیزوزومی و اختلالات میتوکندریایی، هرچند نادر و پیچیدهاند، اما با پیشرفت دانش پزشکی دسترسی به تشخیص دقیق و روشهای کنترل مؤثر فراهم شده است. آگاهی، مشاوره ژنتیک و پیگیری پزشکی منظم، کلید مدیریت موفق این اختلالات و حفظ کیفیت زندگی بیماران است.
منابع:
– Mayo Clinic – Lysosomal storage diseases & Mitochondrial diseases
No.70
۰۴/۰۶